Trimming and assembling
Trimming sequences with Sickle
DNA fastq files from the sequencing machine contain the DNA sequence and the Phred-Q quality scores for each base, whereas fasta files don’t have the quality score. Removing the poorer quality sequence which contains bases with a higher probability of having been misidentified is called trimming, and is necessary for assemblies, mapping and downstream bioinformatics. One program for this is Sickle which takes fastq files and produces trimmed fastq files: two (one of each pair) trimmed sequence files, as well as one trimmed paired reads file. Sickle page on Github
$ sickle pe -f R1.fq -r R2.fq -t sanger -o trimmed_R1.fq -p R2_trimmed.fq -s trimmed_singles_file_R12.fq
 <</font color=”blue”>
<</font color=”blue”>
Note: On terminal, which uses the bash programming language, file extensions don’t matter e.g. my above files R1.fq and R2.fq end in .fq, which shows they are fastq files. They would still be fastq files even if I rename them to R1 and R2. But naming file extensions makes it quicker to see what each file is.
You need to already have Sickle downloaded onto your computer or server, and can open the manual to see the controls using ‘man sickle’ to see what each of the arguments means. I used it for paired files, told it the two fastq input files, the type of sequencing that was used, and what to call the three output files. There are three output files, one for each individual of the paired sequencing read files, and one file where the two pairs are merged together.
The files that Sickle produces are the end are still the giant list of sequences fastq files, but poor quality sequences has been removed. It still keeps the quality score for every base on every read.
Assembling the Contig Map with Spades
After trimming, Spades is a program for assembling small genomes e.g. bacterial and bacteriophage from sequencing reads. It takes every single sequencing read, and looks for shared sequence parts with other sequencing reads to identify overlapping areas to create a contig map. Like Sickle, Spades is also run from the terminal on a mac. It generates a few different output files, so I gave it a new directory to put all the files inside.
$ Spades.py -1 trimmed_R1.fq -2 trimmed_R2.fq -o spades_output_assembly_only --only-assembler
Spades produces lots of output files, including sub-directories called K33, 55, 77 etc. Spades works using an algorithm that looks are sub-strings of different lengths (‘kmers’) of each DNA sequence and looking for matching sub-strings in different DNA sequences to find the overlapping sequences. It repeats this with different length sub-strings to maximise the confidence in the final resulting contig. Read the Spades manual on its website for more details.
Spades also produces the contig sequence, a fasta file. This fasta file has blocks of sequences split into nodes. Spades was finding overlapping DNA sequences by comparing k-mers, and nodes are where many kmers were known to join together to create the long contig map. But maybe the sequencing didn’t cover 100% of the genome, caused by gaps in the sequencing. The sequencing will then end up on different chunks called nodes with unknown gaps between them.
Bandage for seeing node and contig relationships
You can look into your nodes in more depth using Bandage, a program with a graphical output to see how the nodes relate to each other. Spades produces a .fastg output file of nodes which you can give to Bandage (I did this by downloading Bandage onto my computer and clicking, rather than running through terminal.)
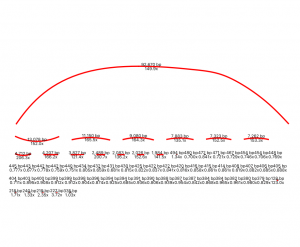
By looking at the graph Bandage produces, I can see I have one larger node/contig, some medium size and lots of smaller ones. I don’t have any bubbles or tangles, so all my DNA sequence are linear and don’t overlap each other.
Bwa mem for aligning DNA sequencing reads onto your new contig
So back onto terminal for running this program. Although Spades was using the DNA sequences to create the contig map, it didn’t keep the information of which DNA read went where. Bwa mem is used to align every DNA sequence back onto your contig map, which at the moment is broken into a multiple nodes/contigs.
The reference genome is your new contig map in its fasta file, and your sequencing files are the trimmed DNA sequences as fastq files from Sickle. The manual can be viewed with man bwa for more information.
Bwa works in two stages, first you need to give it the reference contig so it can create an FM-index to work from. Bwa has different algorithms for doing this depending on sequence lengths, but by not specifying the index type I let bwa pick the most appropriate by itself, which was bwa mem:
$ bwa index scaffolds.fasta
After creating the index, you can align your sequencing reads onto your reference genome by telling it the reference and the two fastq sequencing files:
$ bwa mem scaffolds.fasta -t 16 ../../R1.fq ../../R2.
Bwa mem creates a .bam file which contains information on the contig map, and the depth of coverage for each node.
Converting and Sorting the Bam file with Samtools
Bwa mem created a .sam file of reads aligned to the contig map, but this needs to be a .bam file. Samtools is a program for working with .sam files. My contig map is broken into pieces at the moment, I can sort them by length whilst converting to a .bam file using Samtools, again controlled from the terminal:
$ bwa mem scaffolds.fasta -t 16 ../../R1.fq ../../R2.fq | samtools view -bS -F4 - | samtools sort - -o out.bam
Note: This is 3 steps in one; align the DNA sequences to the contig map with bwa mem, take the output of this, and then sort the output and convert it to a .bam file. The pipe ‘|’ between codes means take the output from out program and put it straight through the next one, using the Bash language for terminal. The -F for Samtools means only DNA sequences which align to the reference genome will be kept.
What to do with your new genome assembly?
Finding and annotating predicted phage genes using Prokka: http://millardlab.org/bioinformatics/lab_server/phage-genome-annotation/
Comparing your new phage genome to other previously published phage genomes: http://millardlab.org/bioinformatics/lab_server/quick-phage-comparison/