A quick comparison of a phage genome against all phage genomes
perl ~/Documents/scripts/run_mash_phage_get_species_JUL18.pl input.fasta
This will compare your phage genome against a database of complete phage genomes using MASH. READ the docs to make sure you know what MASH does. The database is constructed from the monthly download of phage genomes from Genbank. The sketches are precomputed at -s 1000. The output is to screen and produces tab format of
HIT\t QUERY\t DISTANCE\t PROBABILITY\t NUMBER SKETCHES\t TAXA DATA
To redirect to the output to a file use
perl ~/Documents/scripts/run_mash_phage_get_species_JUL18.pl input.fasta >MASH_output.txt
To estimate similarity then 1-Distance(*100) is ~ ANI
MUMMER to compare two genomes
Read the manual so you can work out what the script does
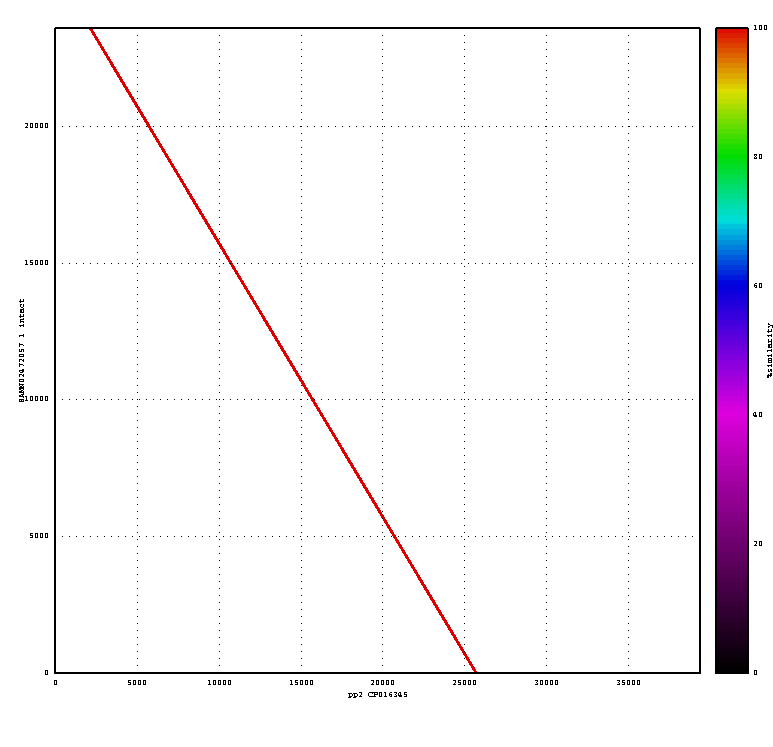
perl ~/Documents/scripts/run_nucmer.pl file1.fa file2.fa
You will get a number of output files. Use
display out.png
To display the ouput of the alignment, which will look like below
All current phage genomes are in /blastdb/Bacteriophage_genomes see http://millardlab.org/bioinformatics/lab_server/ for details of the database.
WARNING the script overwrites existing files of the same name.
For comparing multiple phage genomes at a time use
perl ~/Documents/scripts/run_nucmer_multiple_genomes.pl
or if you wish to use promer then
perl ~/Documents/scripts/run_promer.pl perl/Documents/scripts/run_promer_multiple_gnomes.pl