Ryan has recently been testing the short read eliminator kit from circulomics to enrich for long read for nanopore sequencing of viromes. The input for virome samples was various liquids produced or excreted by cows ….often smelly and sticky, generally not much fun to extract the viral fraction from.
Given the nature of the samples getting large amounts of DNA, let alone HMW DNA from the small sample volumes is not possible. Resorting to MDA amplification to produce enough DNA for the nanopore sequencing. To enrich for longer reads Ryan tried multiple samples without any enrichment with the short read eliminator kit.
The results of which look encouraging
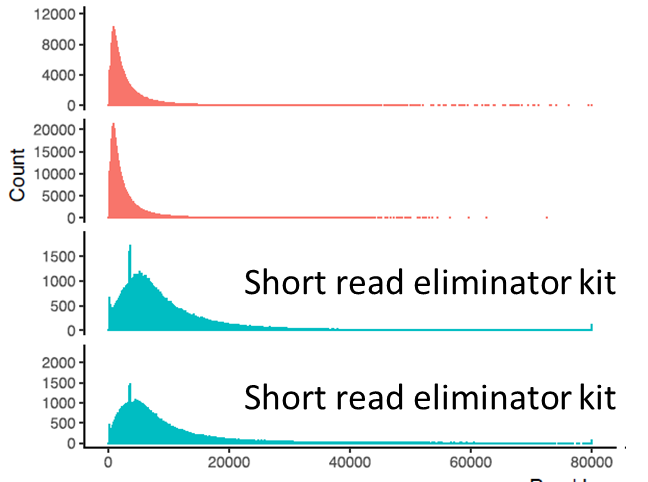
It shifts the median read length from 1.9 kb to 6.9 kb, which still aren’t huge reads. But given average size of phage genomes and the low input issues, it might make a difference to the final assembly. Initial results of assembly of a small number of samples look encouraging, with ~1000 predicted complete phage genomes as predicted from checkV. This is in line with similar number of genomes we have previously assembled from a single seawater sample, where we found a 650 kb phage genome.
We still have to determine how this SRE kit might exclude some phages that have small genomes. As we have previously observed differences in the population of smaller phage genomes when comparing nanopore and illumina sequencing (Cook et al 2021 Microbiome).
*As i found this recently- here is Ryan talking about some of his previous work on PromethION sequencing of viromes