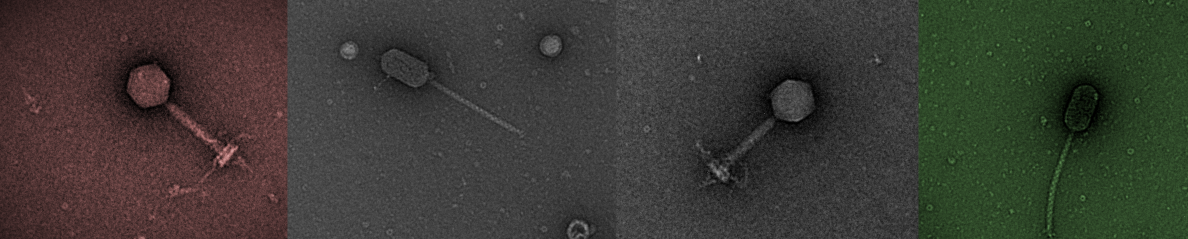
Post assembly a contigs.fasta file will be produced. After mapping reads to all these contigs it should be possible to determine if complete assembly of the phage genome has occurred. If there is a single contig with higher coverage than all other contigs, this suggests this has occured and is a good sign.
To extract the largest contig or contigs above a certain length you can use the script like this
perl ~/Documents/scritps/filter_contigs_on_length.1.pl --file input.fasta --len 10000
This will produced a new file called input_gt10000.fasta that contains all contigs greater than 10000 bp in length.
To extract contigs by name then you can use this script
perl ~/Documents/scripts/filter_contigs_on_name.1.pl --file contigs.fasta --name NODE_1_
This will extract the contig named “>NODE_1_length_92674_cov_68.7486_ID_277” from contigs.fasta and save it to a file called NODE_1_.fasta.